Q&A | 一文读懂沙特SFDA新版产品重大变更指导文件要点及变更解决方案

沙特当地时间3月22日,SFDA发布了新版《医疗器械产品重大与非重大变更的MDMA指导原则》文件。
本次发布的文件是对《Executive Regulation of Law of Medical Devices》章节10-8和《Requirements for Medical Devices Marketing Authorization (MDS-REQ1)》章节5 中对制造商产品存在重大/非重大变更时如何通知或提交报告至沙特SFDA的要求的解释和实例说明。

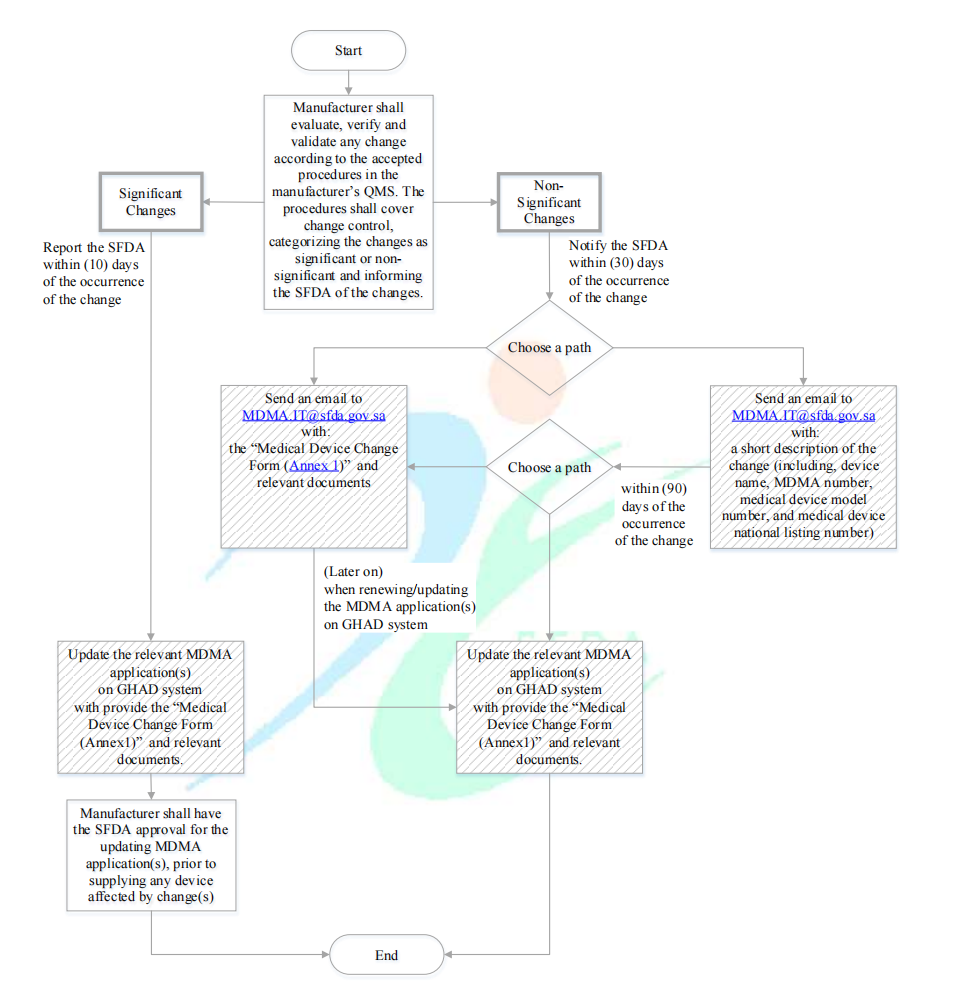
变更提交流程
对于重大变更:变更发生后的10天内向SFDA提交报告(需SFDA审核)。为便于报告的审核,制造商应在GHAD系统上提交相关申请,并提供医疗器械变更申请表(本指导文件附件1提供了模板)和相关说明文档。
对于非重大变更:变更发生后30天向SFDA提交通知(不需要SFDA审核)。为便于通知的备案,制造商应在GHAD系统上提交相关申请。随后向MDMA.IT@sfda.gov.sa发送主题为MDMA证书编号的邮件,邮件附带医疗器械变更申请表(本指导文件附件1提供了模板)和相关说明文档信息。在变更发生后的90天,制造商仍需向MDMA.IT@sfda.gov.sa发送涵盖变更的简短描述的邮件(设备名称、MDMA证书编号、型号和国家目录编号)。
具体操作路径流程图展示如下,
Q&A
为了方便阅读,普瑞纯证也将指导文件中一些问题以Q&A的形式为大家进行整理,

这份文件中提及的重大及非重大变更是如何定义的?
重大变更:
会影响到医疗器械产品的安全、性能的变化,通常有以下呈现:
- 预期用途和标签信息的变更,这些信息被认为是使用该设备的风险缓解措施的重要部分,例如添加禁忌症、警告或其他有关设备安全使用的重要信息;
- 设计方面的变更,例如扩展设备预期用途以包括SFDA未评估的医疗声明,或将预期用户更改为包括新类型的用户;
非重大变更:
不会影响医疗器械产品的安全、性能的变化,通常有以下呈现:
- 添加新的测试验收标准或测试方法,以提供等效或更好的无菌、可靠性等保证;
- 更改包装特性,例如将单个袋装无菌设备放入双层袋中;
- 改变灭菌负载的密度或配置;
- 引入参数释放等质量控制验证和确认过程。

这份文件给到了哪些方面的实例?
本文件列举了诸如以下方面的实例:
- 预期用途和标签的变更;
- 供应商调整;
- 有关质量管理体系的修改;
- 制造工艺、设施或设备的改变;
- 对软件的变更;
- 灭菌方面的调整;
- 产品设计方面的改变;
- 使用材料的变化;
- 安全或性能特性的变更;
更多产品实例分析、匹配和对应解决方案欢迎与我们的咨询顾问进行沟通。
更多有关沙特医疗器械准入内容欢迎点击《中东市场 | 5分钟了解沙特阿拉伯医疗器械注册概况!》查阅。
医疗器械出海产品作为各目的地国强监管之下的商品,一旦涉及变更必然带来大量繁琐的文书、流程及沟通问题,同时均需落实到证书更新环节。普瑞纯证旗下沙特当地自营公司PureKSA拥有丰富的全风险等级医疗器械产品操作经验,按变更次数收费,当地与国内双PM项目全程7X24服务跟进,让您无后顾之忧。
END
普瑞纯证致力于服务中国医疗器械企业扬帆出海,如果各位制造商对中东地区有进一步的需求可以及时与普瑞纯证区域销售专家进行咨询和了解,我们也将会提供不止于多国注册、海外临床等类型的产品。

-普瑞纯证官方咨询顾问-
-获取最新业内资讯-

关于我们
普瑞纯证是行业领先的全球化 SaaS+Data 生命科学服务商,我们的全球服务网络覆盖了包括中国、美国、英国、德国、荷兰、波兰、香港等多个国家和地区。
依托法规认证与临床经验丰富的全球顶尖专家服务团队,普瑞纯证为医疗器械、体外诊断产品等提供全球市场合规准入的全流程咨询服务,以及海外临床试验等一站式解决方案。100+ 国家准入,1000+ 海外注册/认证成功案例,60万+ 全球经销商数据,100万+ 全球临床试验数据,300万+ 全球医械注册数据。从市场战略到法规咨询,助力您的产品全方位顺利合规走向全球市场!