专家解读 | 欧盟委员会发布有关新法规过渡期情况说明指南文件

当地时间7月17日,欧盟委员会公布了一份有关医疗器械产品的Factsheet指南,旨在为(欧盟)境外制造商明确在MDR与IVDR过渡时期需要注意的事项。
普瑞纯证法规专家团队在阅读完更新指南后,结合实际业务推进将就以下更新点与大家予以分享。
过渡期
1
MD产品
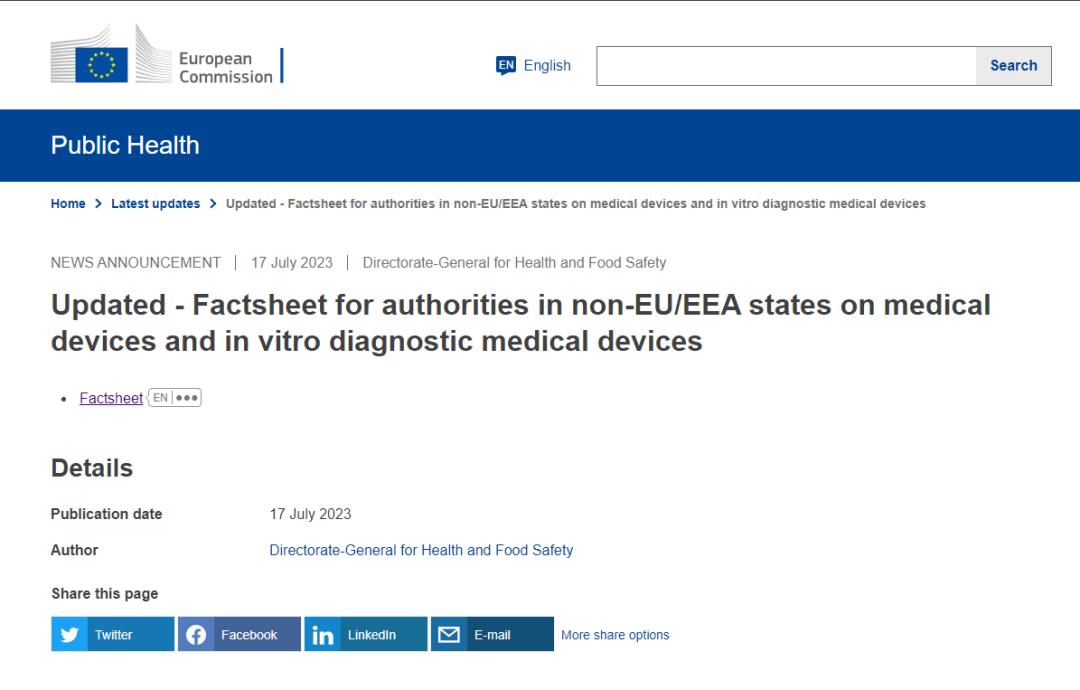
1. 所有1类,首次注册以及具有显著变化的器械,过渡期终点在2021年5月26号,在这之后需要遵循MDR要求,才能在欧盟境内销售;
2. 有显著产品变更(包括设计变更或预期用途变化的)/不符合MDR下QMS要求的/在2024年5月26之前没有递交MDR受理函给公告机构/不再符合MDD或AIMDD要求的/产品现在具有对于健康或安全不可接受的风险的历史遗留器械过渡期终点在2024年5月26,在这只有需要获得MDR证书,才能在欧盟境内销售;
3. 个性化定制植入类产品过渡期终点在2026年5月26号,在这之后需要获得MDR证书,才能在欧盟境内销售;
4. 3类以及2b植入类产品(临床试验豁免产品除外,例如就是缝合线,吻合钉、补牙材料、牙套、牙冠、螺钉、楔子、骨板、线、引脚、夹子和连接器)过渡期终点在2027年12月31号,在这之后需要获得MDR证书才能在欧盟境内销售;
5. 除第4点外的2b类产品、2a、1类灭菌/测量,以及在MDD下不需要公告机构介入但在MDR下需要公告机构介入的产品,过渡期终点在2028年12月31号,在这之后需要获得MDR证书才能在欧盟境内销售。
2
IVD产品
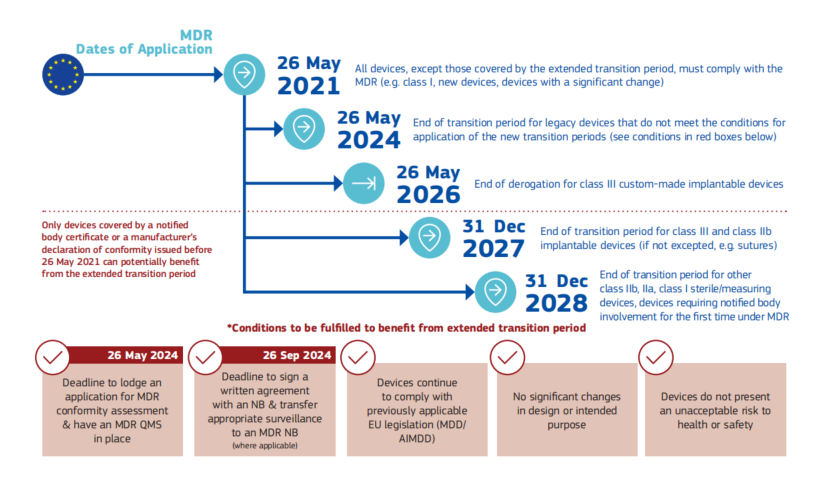
1. 所有Class A非灭菌产品、首次注册以及有显著变化的IVD,过渡期终点是在2022年5月26号,在这之后需要遵循IVDR要求,才能在欧盟境内销售;
2. IVDR下Class D分类产品以及已经获得IVDD证书的产品(例如自测类产品)过渡期终点是在2025年5月26号,在这之后需要获得IVDR证书,才能在欧盟境内销售;
3. IVDR下Class C分类产品过渡期终点是在2026年5月26号,在这之后需要获得获得IVDR证书,才能在欧盟境内销售;
4. IVDR下Class A灭菌类产品以及Class B产品,过渡期终点是在2027年5月26号,在这之后需要获得获得IVDR证书,才能在欧盟境内销售。
历史遗留器械
在过渡期内,所有符合要求的历史遗留器械才能继续在欧盟境内自由销售,此时制造商还需要准备符合过渡期要求的自我声明,关于这份自我声明的要求和DOC是有些不一样,至于模板,欢迎大家添加普瑞客服微信(微信号:purefda)后索取。
除此之外,过渡期间IVDR与MDR历史遗留器械的制造商,还需要注意:
1.最后销售日期。为了保证医疗产品供应链的不会因为法规转换而出现缺货的状态,所谓的“最后销售日期”这个概念是取消的。所有在2021年5月26号之前或在过渡期之内已经在市面销售的MDD/AIMDD产品,均可以一直在市场上流通直到产品有效期过期为止;同样,IVD也是如此,所有在2022年5月26号之前或过渡期之内已经在市面销售的,均可以一直在市场上流通直到产品有效期过期为止;
2. 新规下更加加强了对于临床评估的要求。这点相信所有现在在进行MDR申请的制造商都感受到前所未有的挑战了:过去只需要找同类型产品的相关文献,甚至是同一品牌下,不需要明确规格型号,以及就是简单的可用性报告都能被接受CER。但现在则需要明确等同器械的品牌、规格型号,以及是关联临床文献的准确性才符合要求,过去都能被接受的材料类型,现阶段都会被视为临床证据不充分。
在IVDR申请方面,同样也是如此,大部分产品都被要求需要满足临床试验的评估条件。当前IVD产品还不具备通过临床文献评估以达到豁免临床试验的法规条件,如何通过利用已有资源和合理的路径规划是各位厂商在IVDR申请面临的巨大挑战。
3. 上市后监管
这次我们将重点讨论的是:
Periodic safety update reports安全性周期更新报告:MDR 2a、2b、3类和IVDR C和D类的产品都是需要在获证后,提供PSUR报告的(除这以外的分类,是提供PMS报告)。PSUR报告的模板也有相关指南可以参考:MDCG 2022--21,各位制造商需要注意的是,不同分类的PSUR更新的周期也是不一样的,有些是2年更新即可,有些是需要每年更新。
Trend Report趋势报告:这个说得不多,大家的概念可能不清楚。趋势报告并不属于常规定期更新的报告,它是在制造商发现非严重不良事件的严重程度或发生可能性有增加趋势后,需要撰写的报告。
我们也强烈建议各位存在历史遗留器械的制造商多多注意上市后监管方面的内容。
4. UDI
相关各位厂商在相关产品注册递交公告机构过程中,都需要申请制造商识别码,并且要生成Basic-UDI,同时列名在受理函上,这对于多产品规格型号的制造商也带来了不少的负担。
对于UDI的正式实施期限,整理如下:
MD产品:
1. Class III devices and implantable devices: 26 May 2021
2. Class IIa and class IIb devices: 26 May 2023
3. Class I devices: 26 May 2025
IVD产品
1. Class D devices: 26 May 2023
2. Class B and class C devices: 26 May 2025
3. Class A devices: 26 May 2027
5. EUDAMED数据库
EUDAMED宣称会包含如下6大功能
• Actors registration
• UDI/devices registration
• Notified bodies and certificates
• Clinical investigations and performance studies
• Vigilance and post-market surveillance
• Market surveillance
目前已经可以使用的功能是:Actors registration与 UDI/devices registration。其中Actors registration基本上是强制性使用状态了,因为大家都需要SRN号码,都需要通过此申请。其他功能模块,目前尚未正式开放。
在该Factsheet指南中,指出actors registration、 vigilance and post-market surveillance、clinical investigations and performance studies与market surveillance会在上线后6个月内强制性全面使用;而 UDI/devices registration、Notified Bodies、 certificates会在上线后24个月内强制性全面使用。
我们也希望能看到EUDAMED能有更多的公开数据信息,以提升监管透明度。
以上就是Factsheet指南的内容了,主要还是说了很多历史遗留器械的注意事项,希望对各位制造商有所帮助。如果您的相关产品在前往欧盟地区存在准入等需求,欢迎与我们的销售顾问进行沟通。


-普瑞纯证官方咨询顾问-
-获取最新业内资讯-

关于我们
普瑞纯证是行业领先的全球化AI赋能生命科学服务商,我们的全球服务网络覆盖了包括中国、美国、英国、德国、荷兰、波兰、香港等多个国家和地区。
依托法规认证与临床经验丰富的全球顶尖专家服务团队,普瑞纯证为医疗器械、体外诊断产品等提供全球市场合规准入的全流程咨询服务,以及海外临床试验等一站式解决方案。100+ 国家准入,1000+ 海外注册/认证成功案例,60万+ 全球经销商数据,100万+ 全球临床试验数据,500万+ 全球医械注册数据。从市场战略到法规咨询,助力您的产品全方位顺利合规走向全球市场!