深度解读 | 制造商请注意!MDR过渡期延长更新,旧证书有效期不再考虑!

一个月前,有相关议案出台指出MDD&AIMDD到MDR的过渡期可能会延长。当时就有客户咨询过普瑞君,具体是以旧证书的有效期,还是以议案的日期为准,普瑞君当时的回答是,这要看后续发布法案如何定义。
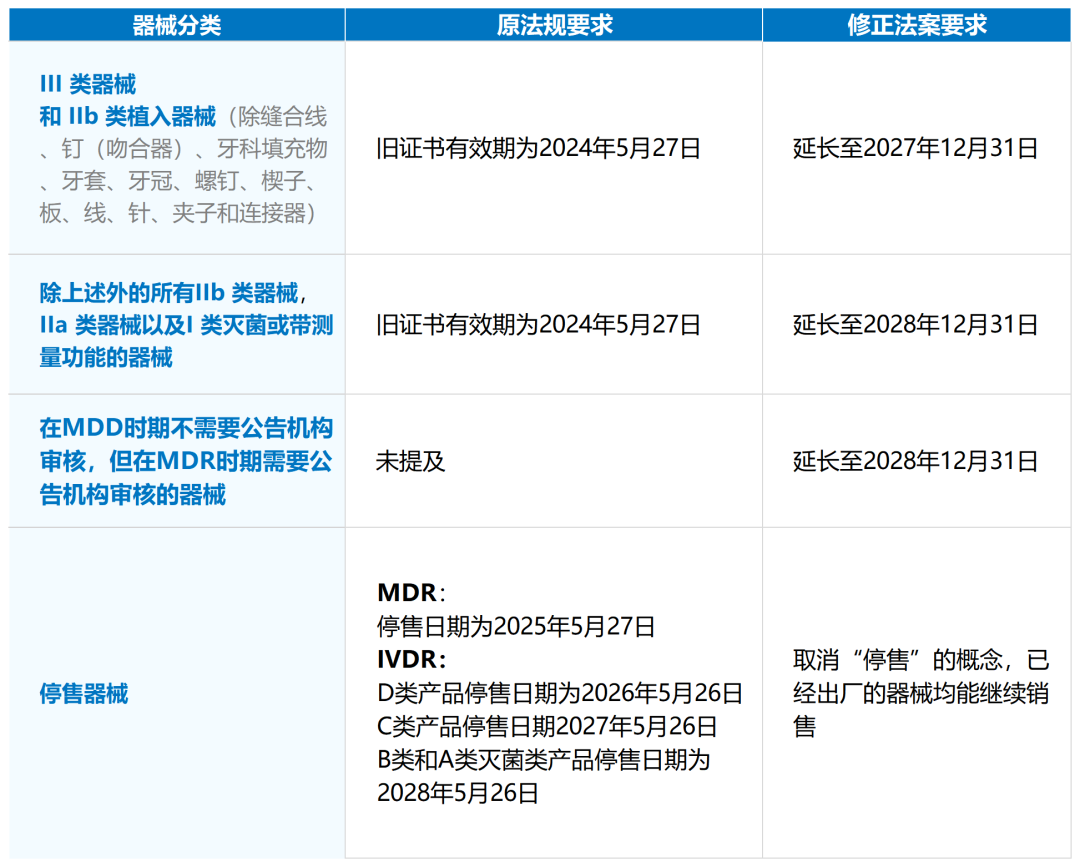
欧洲当地时间2023年1月6日,关于MDR过渡期延长的提案通过,确定了相关问题的答案,细节如下:
重点解读
1.关于停售问题
该法案总体针对医疗器械,但关于“停售”产品,涵盖了IVD与MD两者,同时修正了MDR法规与IVDR过渡期延长法案(可以理解成IVDR Article 110法规修正再修正,欧盟在延长过渡期这件事上频繁更新,后续可能还有类似动作)。在该修正法案中也解释了为什么会取消最后销售日期的原因:为了避免有效、安全的医疗器械的无谓浪费。
2. 关于适用器械
和IVDR过渡期延长法案一样,该法案针对的都是遗留器械(Legacy device),对于那些以前是没有MDD或AIMDD证书的医疗器械,可以忽略该红利了。
3. 关于旧证书有效期与过渡期终止日期
法规原Article 120有两个时间概念:旧证书有效期与过渡期终止日期;在修正法案中,不再考虑旧证书有效期,只要符合条件的遗留器械(Legacy device),统一以“过渡期终止日期”为准。
4. 该修正法案和IVDR过渡期延长法案最大不同
在旧法规证书有效期或最迟在2024年9月26日之前,制造商需要与公告机构签订审核合同,才能获得过渡期延长的红利。这点在IVDR过渡期延长法案是没有要求的。当然除了与公告机构签订审核合同外,还有别的法子,就是和欧盟成员国主管当局获得审核废除许可,不过这条可行性基本上为零了,就不展开讨论了。
说回可行性最高的与公告机构签订审核合同这点,还有一个细节需要注意:法案中明确制造商最迟需要在2024年5月26日前向公告机构提出申请,然后最迟是在2024年9月26日前双方签订审核合同。此外,修正法案上没有明确是否是要与原证书的公告机构,也没有明确是否只限定一家,但是很清晰的是:不是受理函,是签订审核合同(这就意味着通常要预缴款50%)。所以可以理解成,产品是计划在哪家公告机构审核MDR,就与哪家公告机构签订合同。当然,土豪请随意。
5.关于遗留器械(Legacy device)
遗留器械(Legacy device) 不能有产品设计上、预期用途上的显著变化。那些连预期用途都变化的产品,制造商自己要先想清楚是否合适该修正法案了,普瑞君不多说了,叫不醒装睡的人。
在2024年5月26日前,所有遗留器械(Legacy device)质量体系建设必须是符合MDR下的EN ISO 13485:2016。MDR法规下,对于新增了很多对质量体系的要求,例如新增了临床评估程序、PMCF程序、UDI程序等,这些质量体系的建设,制造商们都最迟要在2024年5月26日前完成。其实现在年审,已经越来越多制造商被收到了上市后监管文件不足的发补了,但是也不是全面MDR法规要求下的质量体系要求,预计未来年审时,针对质量体系这块,审核员的要求会逐渐向MDR Article 10 看齐。
总体来说,最重要也是最难的一点就是如何与公告机构签订MDR审核合同,只有在规定时间前(旧证书有效期内或在2024年9月26日前签订)才能享受到过渡期延长的红利。证书即将过期的制造商们在这个时候,究竟是否能够又快又好联系公告机构资源完成从受理到获批的全流程工作,就显得尤为重要。普瑞纯证与欧洲多家公告机构达成战略合作关系,目前已经推出技术文档提交和受理函申请业务,有需要的制造商都可以联系我们的专家顾问团队哦~

-普瑞纯证官方咨询顾问-
-获取最新业内资讯-

关于我们
普瑞纯证是行业领先的全球化 SaaS+Data 生命科学服务商,我们的全球服务网络覆盖了包括中国、美国、英国、德国、荷兰、波兰、香港等多个国家和地区。
依托法规认证与临床经验丰富的全球顶尖专家服务团队,普瑞纯证为医疗器械、体外诊断产品等提供全球市场合规准入的全流程咨询服务,以及海外临床试验等一站式解决方案。100+ 国家准入,1000+ 海外注册/认证成功案例,60万+ 全球经销商数据,100万+ 全球临床试验数据,300万+ 全球医械注册数据。从市场战略到法规咨询,助力您的产品全方位顺利合规走向全球市场!